Influence of mild traumatic brain injury during pediatric stage on short-term memory and hippocampal apoptosis in adult rats
Article information
Abstract
Traumatic brain injury (TBI) is a leading cause of neurological deficit in the brain, which induces short- and long-term brain damage, cognitive impairment with/without structural alteration, motor deficits, emotional problems, and death both in children and adults. In the present study, we evaluated whether mild TBI in childhood causes persisting memory impairment until adulthood. Moreover, we investigated the influence of mild TBI on memory impairment in relation with hippocampal apoptosis. For this, step-down avoidance task, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay, and immunohistochemistry for caspase-3 were performed. Male Sprague-Dawley rats were used in the experiments. The animals were randomly divided into two groups: sham-operation group and TBI-induction group. The mild TBI model was created with an electromagnetic contusion device activated at a velocity of 3.0 m/sec. The results showed that mild TBI during the pediatric stage significantly decreased memory retention. The numbers of TUNEL-positive and caspase-3-positive cells were increased in the TBI-induction group compared to those in the sham-operation group. Defective memory retention and apoptosis sustained up to the adult stage. The present results shows that mild TBI induces long-lasting cognitive impairment from pediatric to adult stages in rats through the high level of apoptosis. The finding of this study suggests that children with mild TBI may need intensive treatments for the reduction of long-lasting cognitive impairment by secondary neuronal damage.
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of neurological deficit in brain, which induces short- and long-term brain damage, cognitive impairment with/without structural alteration, motor deficits, emotional problems, and death both in children and adults (Langlois and Sattin, 2005; Levine et al., 2008; Margulies, 2000; Milman et al., 2005; Tashlykov et al., 2007). The symptoms of TBI can be divided into primary and secondary injuries. The primary injuries results immediately from direct mechanical impact, while the secondary injuries are induced by delayed pathological tissue reaction, including apoptotic mechanisms within hours or months following the primary injury (Bayir et al., 2003; Park et al., 2007; Shaw, 2002; Zhang et al., 2008).
The main problem associated with TBI is that many people suffer cognitive dysfunction such as memory deficits after TBI (Greve et al., 2003). There is a possibility that children with TBI in particular, may end up with persistent medical, cognitive and behavioral sequelae (Anderson et al., 2005; Ewing-Cobbs et al., 2006). This is because the immature brain is less resistant to mechanical forces which cause displacement of brain tissues as a results of inadequate protection due to insufficient calcification of the skull (Bauer and Fritz, 2004). However, it is difficult to determine the relationship between children’s and adults’ TBI (Carbonell and Grady, 1999; Runnerstam et al., 2001), because the majority of children’s TBI is often classified as mild TBI which produces cognitive deficits and behavioral changes without apparent structural damage (Bazarian et al., 2006; Milman et al., 2005; Roe et al., 2009; Zohar et al., 2003).
Although the morphological and physiological pathologies which underline mild TBI are not yet fully understood, several studies have suggested that apoptosis is a key mechanism that is involved in secondary or delayed neuronal cell death (Povlishock, 2000; Tehranian et al., 2008; Zhang et al., 2008). It has also been reported that the hippocampus and corpus callosum are primarily vulnerable regions for significant long-term structural and volume alteration by TBI (Kim et al., 2010; Tomaiuolo et al., 2004). For this reason, most studies suggest neuronal cell damage or cell death of axon as well as cell bodies in the brain as the possible mechanism for the mild TBI-induced cognitive impairment (Bayir et al., 2003; Levine et al., 2008; Reeves et al., 2005; Runnerstam et al., 2001; Singleton and Povlishock, 2004; Tehranianet al., 2008; Zhang et al., 2008).
In the present study, we evaluated whether mild TBI in childhood causes persisting memory impairment until adulthood (Levine et al., 2008; Povlishock, 2000; Vakil, 2005). Moreover, we investigated the influence of mild TBI on memory impairment in relation with hippocampal apoptosis. For this, step-down avoidance task, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay, and immunohistochemistry for caspase-3 were performed.
MATERIALS AND METHODS
Animals and treatments
The experimental procedures were conducted in accordance with the animal care guidelines of the National Institutes of Health (NIH) and the Korean Academy of Medical Sciences. Male Sprague-Dawley rats weighing 90±10 g (4 weeks of age) were used in the experiments. The rats were housed under controlled temperatures (20±2°C) and lighting conditions (07:00 to 19:00), with food and water made available ad libitum throughout the experiments. The animals were randomly divided into two groups (n =40 in each group): the sham-operation group and the TBI-induction group.
Surgical procedure of mild TBI
The surgical procedures were performed using aseptic procedures and conditions. The TBI model used in the present study was based on a previous study (Gavrieli et al., 1992). The rats were anesthetized using Zoletil 50® (10 mg/kg, i.p.; Vibac Laboratories, Carros, France) and prepared for surgery. When a rat was unresponsive (without ocular or pedal reflexes), the head was shaved and placed into a digital stereotaxic device (stereotaxic frame, Benchmark DeluxeTM; MyNeurolab, St. Louis, MO, USA). A circular craniotomy (5.0 mm) was performed using a Dremel motor tool and a specially designed drill that prevented damage to the meninges and cortex (2.4 mm lateral to the midline and 4.2 mm posterior to the coronal suture). The contusion injury was created with an electromagnetic contusion device (Impact One™, Stereotaxic Impactor; MyNeurolab) using a sterile, stainless steel impactor tip (3.0 mm diameter) that was activated at a velocity of 3.0 m/sec. The impactor tip positioned on the cortex and resulted in 2.5 mm compression on the cortex. Following the contusion, any bleeding was controlled with sterile sponges soaked in cold saline, and the incisions were closed with nylon suture material. The rats in the sham operation group were treated in an identical manner; however, brain contusions were not administered.
Step-down avoidance task
In order to evaluate the short-term memory of rats, the latency in the step-down avoidance task was conducted as the described previously method (Izquierdo et al., 1995). On the 7 days after induction of TBI, the rats were trained in a step-down avoidance task for 2 h. After training, the latency (sec) of the animals in each group was determined. The rats were positioned on a 7×25 cm platform with a height of 2.5 cm, and were allowed to rest on the platform for 2 min. The platform faced a 42 cm×25 cm grid of parallel 0.1 cm-caliber stainless steel bars, which were spaced 1 cm apart. During the training sessions, the animals received a 0.5 mA scramble foot shock for 20 sec immediately upon stepping down. Retention time was assessed on the 7 days, 4 weeks, 8 weeks, and 12 weeks after induction of TBI. The interval of rats stepping down and placing all four paws on the grid was defined as the latency in the step-down avoidance task. Any latency over 180 sec was counted as 180 sec.
Tissue preparation
For brain tissue preparation, the rats were sacrificed on the 30 min, 6 h, 24 h, 48 h, 7 days, 4 weeks, 8 weeks, and 12 weeks after induction of TBI. In brief, the animals were fully anesthetized using Zoletil 50® (10 mg/kg, i.p.; Vibac Laboratories, Carros, France), transcardially perfused with 50 mM phosphate-buffered saline (PBS), and then fixed with 4% paraformaldehyde in 100 mM phosphate buffer (PB, pH 7.4). The brains were dissected, postfixed in the same fixative overnight, and finally transferred into a 30% sucrose solution for cryoprotection. Coronal sections of 40 μm thickness in each section of the brain was made with a freezing microtome (Leica, Nussloch, Germany).
TUNEL staining
To visualize DNA fragmentation, TUNEL staining was performed using an In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany) according to the manufacturer’s protocol (Gavrieli et al., 1992; Jin et al., 2014). The sections were postfixed in ethanol–acetic acid (2:1), rinsed and then incubated with proteinase K (100 μg/ml). They were then rinsed and incubated in 3% H2O2, permeabilized with 0.5% Triton X-100, rinsed again, and incubated in the TUNEL reaction mixture. Finally, they were rinsed and visualized using a Converter-POD with 0.03% 3,3′-diaminobenzidine (DAB). Mayer’s hematoxylin (DAKO, Glostrup, Denmark) was used as a counter stain, and the sections were mounted onto gelatin-coated slides. The slides were air-dried overnight at room temperature, and cover slips were mounted using Permount®.
Immunohistochemistry for caspase-3
An immunohistochemistry was conducted to evaluate the caspase-3 expressions in the hippocampus, according to the previously described method (Jin et al., 2014; Sim et al., 2004). In brief, an average of 10 sections within the hippocampus spanning from Bregma −2.30 mm to −4.30 mm were obtained from each brain. The selected regions were all of a uniform hippocampal shape. Staining was carried out using free-gloating sections. The sections were incubated in PBS for 5 min and washed three times in the same buffer. They were then incubated in 1% hydrogen peroxide (H2O2) for 30 min to block the endogenous peroxidase activity. After washing in PBS, the sections were incubated in blocking serum (10% normal horse serum and 0.1% Triton X-100 in PBS) for 2 h. Next, they were incubated overnight with caspase-3-specificmouse monoclonal antibody solution (1:500; Santa Cruz Biotechnology, Santa Cruz, CA). Once more, they were incubated for 2 h with the biotinylated anti-mouse secondary antibody (1:200; Vector Laboratories, Burlingame, CA, USA). Next, the sections were incubated with avidin-biotin-peroxidase complex (1:100; Vector Laboratories) for 1 h at room temperature. For staining, they were incubated in a solution consisting of 0.02% 3,3’-diaminobenzidine tetrahydrochloride (DAB) and 0.03% H2O2 in 50 mM Tris-hydrochloride (HCl) (pH 7.6) for approximately 3 min, after which they were washed with PBS and mounted onto gelatin-coated slides. The slides were air-dried overnight at room temperature, and cover slips were mounted using Permount®. The number of Caspase-3-positive cells was counted hemilaterally, with a light microscope at ×400 magnification (Olympus, Tokyo, Japan), and the area in the selected region of the hippocampus was measured using the Image-Pro® Plus software (Media Cybernetics, Silver Spring, MD, USA).
Statistical analysis
Data analysis was preformed with SPSS 16.0 software. The differences between the two groups were analyzed with t-test, whereas the differences between multiple groups were analyzed with ANOVA test and Duncan’s post-hoc test. Two-sided P<0.05 was considered significant. All values were expressed as the mean ±standard error of the mean (SEM).
RESULTS
The effect of mild TBI during the pediatric stage on the short-term memory in the adult rats
The latencies of the step-down avoidance task are presented in Fig. 1. The mean latency in both the sham-operation group and TBI-induction group was 269.42±10.26 sec before the induction of TBI. In the sham-operation, the mean latency was 195.14±14.15 sec at 7 days, 229.00±13.55 sec at 4 weeks, 235.00±16.45 sec at 8 weeks, and 224.83±17.92 sec at 12 weeks. In the TBI-induction group, the mean latency was 87.0±13.72 sec at 7 days, 134.14±15.98 sec at 4 weeks, 135.14±15.05 sec at 8 weeks, and 143.42± 14.41 sec at 12 weeks. These results showed that mild TBI during the pediatric stage significantly decreased memory retention compared to that in the sham-operation group, and that this defective memory retention sustained until to the adult stage.
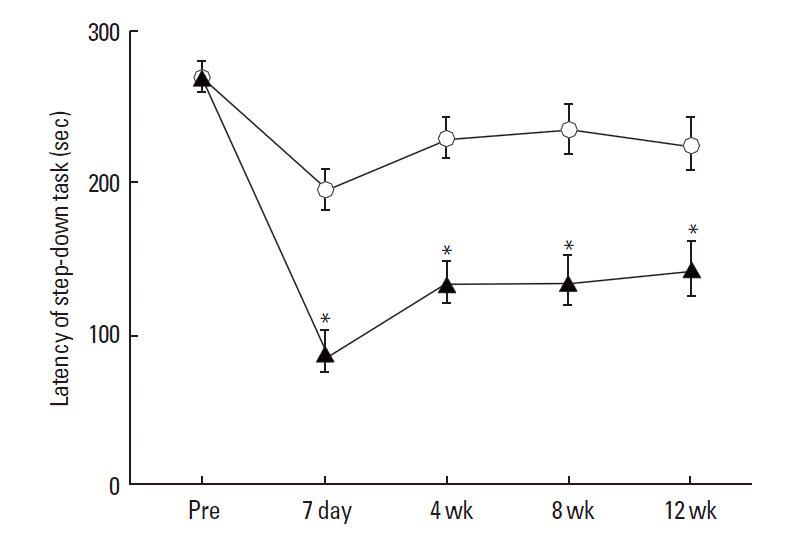
The effect of mild traumatic brain injury (TBI) during the pediatric stage on latency in the step down avoidance task in the adult stage of rats. (○) Sham-operation group, (▴) TBI-induction group. The data present means± standard error of the mean. *Represents P< 0.05 compared to the sham-operation group in each time.
The effect of mild TBI during the pediatric stage on the number of TUNEL-positive cells in the hippocampus of adult rats
Photomicrographs of TUNEL-positive cells in the dentate gyrus are presented in Fig. 2. In the sham-operation group, the mean number of TUNEL-positive cells in the dentate gyrus was 8.161±96.30/mm2 at 30 min, 12.72±2.62/mm2 at 6 h, 14.09± 2.82/mm2 at 24 h, 12.85±4.34/mm2 at 48 h, 7.50±1.89/mm2 at 7 days, 9.75±1.80/mm2 at 4 weeks, 13.59±2.55/mm2 at 8 weeks and 18.26±3.32/mm2 at 12 weeks.
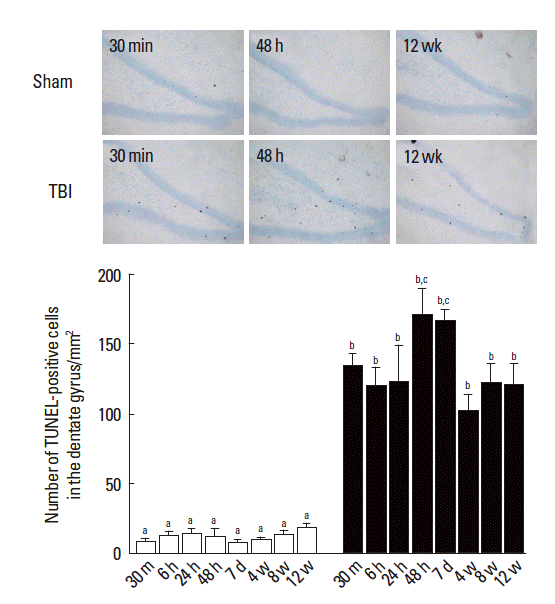
The effect of traumatic brain injury (TBI) during the pediatric stage on TUNEL-positive cells in adult hippocampus. Upper: photomicrographs of TUNEL-positive cells in the dentate gyrus of hippocampus. The scale bar represents 50 μm. Lower: number of TUNEL-positive cells in the dentate gyrus by time. The data present means± standard error of the mean. Symbols a, b, c represents statistical significance (P< 0.05) in each group and time.
In the TBI-induction group, the mean number of TUNEL-positive cells in the dentate gyrus was 135.43±6.89/mm2 at 30 min, 119.58±13.06/mm2 at 6 h, 123.48±24.69/mm2 at 24 h, 171.53±17.12/mm2 at 48 h, 166.84±6.98/mm2 at 7 days, 102.46± 11.65/mm2 at 4 weeks, 122.97±12.46/mm2 at 8 weeks, and 120.72±14.34/mm2 at 12 weeks. These results showed that mild TBI during the pediatric stage significantly increased the mean number of TUNEL-positive cells in the dentate gyrus compared to that in the sham-operation group at each measurement time, and this mild TBI-induced increment of TUNEL-positive cells sustained in the dentate gyrus until the adult stage of rats.
The effect of mild TBI during the pediatric stage on caspase-3-positive cells in the hippocampus of adult rats
Photomicrographs of caspase-3-positive cells in the dentate gyrus are presented in Fig. 3. In the sham-operation group, the mean number of caspase-3-positive cells in the dentate gyrus was 19.72±3.86/mm2 at 30 min, 20.24±2.60/mm2 at 6 h, 29.95± 6.51/mm2 at 24 h, 30.80±5.00/mm2 at 48 h, 30.20±3.29/mm2 at 7 days, 33.14±4.82/mm2 at 4 weeks, 37.09±5.08/mm2 at 8 weeks, and 32.53±5.19/mm2 at 12 weeks.
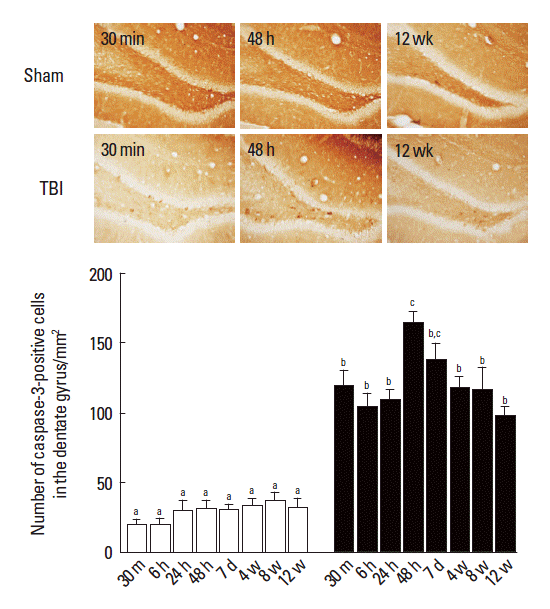
The effect of traumatic brain injury (TBI) during the pediatric stage on caspase-3-positive cells in the adult hippocampus. Upper: photomicrographs of caspase-3-positive cells in the dentate gyrus of hippocampus. The scale bar represents 50 μm. Lower: number of caspase-3-positive cells in the dentate gyrus by time. The results present means± standard error of the mean. Symbols a, b, c represents statistical significance (P< 0.05) in each group and time.
In the TBI-induction group, the mean number of caspase-3-positive cells in the dentate gyrus was 119.19±9.84/mm2 at 30 min, 104.74±8.77/mm2 at 6 h, 109.63±5.94/mm2 at 24 h, 164.44± 6.62/mm2 at 48 h, 137.75±11.85/mm2 at 7 days, 117.82±7.17/mm2 at 4 weeks, 116.63 ±15.23/mm2 at 8 weeks, and 97.67±5.69/mm2 at 12 weeks. These results showed that mild TBI during the pediatric stage significantly increased the mean number of caspase-3-positive cells in the dentate gyrus compared to that in the sham-operation group at each measurement time, and this mild TBI-induced increment of caspase-3-positive cells sustained in the dentate gyrus until the adult stage of rats.
DISCUSSION
The present results demonstrate that cognitive impairment by mild TBI during the pediatric stage lasts up to adulthood. It is well known that TBI results in cognitive deficit and memory impairment in humans and animal models (Vakil, 2005). Several studies have reported that minimal or mild TBI also showed long-term behavioral and cognitive deficits (Raghupathi et al., 2000; Tashlykov et al., 2007; Zohar et al., 2003). Zohar et al. (2003) showed that injured mice could not improve their escape latency more than 50% of their initial performance. This means that even minimal impact on the brain might cause persistent memory deficit in adult mice. Tashlykov et al. (2007) also suggested that minimal brain trauma in adult mice is associated with cognitive and behavioral deficits. In the present study, we demonstrated that mild TBI in the childhood stage impaired memory retention in step-down avoidance task, and this defective memory retention by mild TBI lasts up to adulthood. The present result revealed that short-term memory impairment by mild TBI in the childhood stage sustained through the adulthood.
Although the exact mechanism of TBI-induced cognitive impairment is not clear, several pathological factors, including direct neuronal cell loss, traumatic axonal injury, hemorrhage, ischemia, changes of brain blood barrier (BBB) permeability, and long-term structural alteration have been proposed (Gennarelli and Graham, 1998; Levine et al., 2008; Povlishock, 2000; Reeves et al., 2005; Singleton and Povlishock, 2004). On the other hand, some studies suggest that cognitive impairment by mild TBI is not associated with the BBB permeability change, neurological injury, or apparent structural damage by direct mechanical force. It is relevant to the persistent apoptotic process or alteration in synaptic plasticity by primary injury-induced secondary neurochemical events (Albensi 2001; Bazarian et al., 2006; Graham et al., 2000; Milman et al., 2005; Raghupathi et al., 2000; Roe et al., 2009; Zhang et al., 2008; Zohar et al., 2003).
In the present study, mild TBI increased the expressions of apoptotic changes, such as TUNEL-positive and caspase-3-positive cells, in the hippocampus compared to the young rats in the sham-operation group. The enhancement of hippocampal apoptosis by mild TBI peaked at 48 h and lasts for 12 weeks. Similar to our findings, previous studies showed that apoptosis was initiated at 12–72 h after TBI (Beer et al., 2001; Johnson et al., 2005), and these apoptotic changes were found to up to a month (Sato et al., 2001). Mild TBI could induce undiagnosed and/or diffused neuronal damages and apoptosis in multiple brain areas, including the hippocampus, without visible structural damages in human adults (Bigler et al., 2002; Raghupathi et al., 2000). Delayed and/or diffused neuronal damage by closed minimal TBI on head attributed sustained cognitive impairment in adult mice (Tashlykov et al., 2007). Moreover, the development of neuronal cell loss is not the only result of mechanical impact, but it is also a secondary, delayed pathological tissue reaction (Zhang et al., 2008). The possibility that cognitive and behavioral deficits in childhood TBI may be persistent has been suggested (Anderson et al., 2005; Bauer and Fritz, 2004; Ewing-Cobbs et al., 2006). Based on the present results, it is possible that the maintenance of increased apoptosis after mild TBI causes the sustained memory deficits from the pediatric stage up to the adult stage.
In conclusion, mild TBI induces long-lasting cognitive impairment from pediatric to adult stages in rats through the high level of apoptosis. The finding of this study suggests that children with mild TBI may need intensive treatments for the reduction of long-lasting cognitive impairment by secondary neuronal damage.
Notes
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2011-0021908).